
Presentación de Caso
Amiloidosis de mesenterio y vasos sanguíneos
Amyloidosis of the mesentery and blood vessel
José
Fernández Sotolongo1* https://orcid.org/0000-0003-1587-5443
Yosvani
Josué Ortiz Montero1 https://orcid.org/0000-0001-6049-2375
Miguel
Bernal Duran1 https://orcid.org/0009-0009-7667-307X
Licet
González Fabián2 https://orcid.org/0000-0003-0466-7251
1Hospital
Universitario "General Calixto García". Universidad de Ciencias
Médicas de la Habana. Facultad "Calixto García". La
Habana, Cuba.
2Instituto
de Gastroenterología. La Habana, Cuba.
*Autor para la correspondencia: jose.fernandez@infomed.sld.cu
RESUMEN
Introducción:
La amiloidosis es una enfermedad de etiología desconocida, que se caracteriza
por el depósito de sustancia amorfa (amiloide), en los espacios extracelulares
de diversos órganos y tejidos, produce alteraciones funcionales y estructurales
según la localización e intensidad del depósito.
Objetivo: Describir las alteraciones clínicas, de laboratorio
e imagenológicas en un paciente con amiloidosis de mesenterio.
Caso clínico: Paciente masculino de 43 años de edad, raza
blanca con antecedentes de salud, refiere que hace un año comenzó
con diarreas liquidas, en número de 5 a 6 por día, sin flemas
ni sangre, acompañado de dispepsia, astenia, adinamia, edemas en ambos
miembros inferiores y pérdida de peso de 30 kilogramos. Al examen físico
como hallazgo positivo el tejido celular subcutáneo infiltrado en ambos
miembros inferiores hasta nivel de las rodillas, grado II, de difícil
godet. Se ingresó para estudio en sala de gastroenterología, se
le realizaron ecografía abdominal y tomografía axial computarizada
de abdomen con contraste oral y endovenoso con hallazgos compatibles con paniculitis
mesentérica. Panendoscopia oral que diagnosticó duodenitis eritematosa
nodular. Biopsia de mesenterio con diagnóstico histológico compatible
con amiloidosis de mesenterio. Al paciente se le brinda atención multidisciplinaria
por parte de Gastroenterología, Nefrología, Hematología
y Cardiología con tratamiento personalizado y una evolución favorable.
Conclusiones: La amiloidosis es un diagnóstico infrecuente, que
no existe un protocolo de tratamiento de aceptación universal, requiere
de una atención médica multidisciplinaria con un tratamiento específico
y personalizado en dependencia de la extensión y estadío evolutivo
de la enfermedad.
Palabras clave: Amiloidosis, mesenterio, vasos sanguíneos.
ABSTRACT
Introduction:
Amyloidosis is a disease of unknown etiology, characterized by the deposition
of amorphous substance (amyloid) in the extracellular spaces of various organs
and tissues, causing functional and structural alterations depending on the
location and intensity of the deposition.
Objective: To describe the clinical, laboratory, and imaging alterations
in a patient with a rare diagnosis of mesenteric amyloidosis.
Clinical case: A 43-year-old white male patient with a background of
good health reports that a year ago he began experiencing liquid diarrhea, 5
to 6 times per day, without phlegm or blood, accompanied by dyspepsia, asthenia,
adynamia, edema in both lower limbs, and a weight loss of 30 kilograms. On physical
examination, a positive finding was subcutaneous tissue infiltration in both
lower limbs up to the knees, grade II, with difficult pitting edema. He was
admitted for study in the gastroenterology ward; an abdominal ultrasound and
abdominal computed tomography with oral and intravenous contrast were performed,
showing findings compatible with mesenteric panniculitis. Nodular erythematous
duodenitis was diagnosed after performing oral panendoscopy. Mesenteric biopsy
revealed histological diagnosis compatible with mesenteric amyloidosis. The
patient receives multidisciplinary care from Gastroenterology, Nephrology, Hematology,
and Cardiology with personalized treatment and a favorable evolution.
Conclusions: Amyloidosis is an uncommon diagnosis, for which there is
no universally accepted treatment protocol. It requires multidisciplinary medical
care with specific and personalized treatment depending on the extent and evolutionary
stage of the disease.
Keywords: Amyloidosis, mesentery, blood vessels.
Recibido:
21/02/2026
Aprobado: 20/04/2026
INTRODUCCIÓN
La amiloidosis es una enfermedad infiltrante, sistémica o limitada a un órgano que resulta del depósito en los tejidos de la proteína amiloide (una proteína fibrilar resistente a la proteólisis con cadena B - plegada como estructura secundaria. El amiloide es una glucoproteína compleja, eosinofílica e insoluble que muestra birrefringencia verde con la luz polarizada, luego de la tinción con rojo Congo. Las manifestaciones clínicas son inespecíficas, determinadas por el órgano o sistema afectado. Los sitios principales donde se deposita el amiloide en el intestino son las paredes de los vasos sanguíneos y la muscular de la mucosa donde deteriora la absorción.(1)
En el siglo XIX los padres de la patología moderna Rudolf Virchow y Carl F. Rokitansky, observaron en autopsias un proceso infiltrativo en los distintos órganos que denominaron degeneración lardácea (del latín, laridum que significa semejante a la manteca); al conservar dichos órganos en yodo, se comprobó que adquirían una coloración azul semejante al almidón, fue así como se comenzó a utilizar el término amilosis o amiloidosis (del griego Amylon, almidón).(2)
La incidencia de amiloidosis es difícil de definir. En Estados Unidos ajustada a la edad es estimada entre 5,1 y 12,8 por millón de personas por año, el 60 % de los pacientes diagnosticados entre 50 y 70 años de edad. En Occidente predomina la Amiloidosis de cadena ligera (AL) con una incidencia de 10 a 12 casos por millón de personas al año con una mediana de edad al diagnóstico de 63 años y un predominio en el sexo masculino.(3)
Existen varios subtipos de amiloidosis con diferentes unidades estructurales de proteína amiloidea. Estas incluyen amiloidosis primaria (amiloidosis de cadenas ligeras), amiloidosis secundaria (amiloidosis por amiloide A) y amiloidosis familiar. Las unidades estructurales de la proteína amiloidea para estos tres subtipos son fragmentos de cadenas ligeras de inmunoglobulina monoclonal, producto de interrupción de una proteína reactante de fase aguda conocida como amiloide A sérico y varias proteínas mutantes. Puede producir síntomas gastrointestinales desde la boca hasta el ano, aunque la intestinal puede ser asintomática.(4)
El diagnóstico se establece por el examen histológico. La biopsia rectal determina el diagnóstico en alrededor del 80 %, datos recientes indican que la biopsia de duodeno puede ser positivas en el 100 %. La biopsia del panículo de grasa abdominal es segura y diagnóstica.(5)
No hay un tratamiento específico para la amiloidosis, depende de cada caso, de la extensión y el estadio evolutivo de la enfermedad.(5)
El objetivo de la actual presentación es describir las alteraciones clínicas, de laboratorio e imagenológicas en un paciente con amiloidosis de mesenterio.
PRESENTACIÓN DEL CASO
Paciente masculino de 43 años de edad, raza blanca con antecedentes de salud. Acude a consulta porque presenta desde hace un año diarreas liquidas, en número de 5 a 6 por día, sin flemas ni sangre, acompañado de dispepsia, astenia, adinamia, edemas en ambos miembros inferiores y pérdida de peso de 30 kilogramos. Se le realizó examen físico en el que se observó como hallazgo positivo: tejido celular subcutáneo infiltrado en ambos miembros inferiores hasta nivel de las rodillas, grado II, de difícil godet.
Se le realizaron complementarios de laboratorio clínico e imagenológicos que reportaron como hallazgos positivos:
La ecografía abdominal informó hepatomegalia homogénea, diámetro mayor 17 cm, aumento de la ecogenicidad de 2 centímetros, la vesícula biliar con múltiples litiasis de pequeño tamaño. Porta dilatada en 17 mm, flujo hepatopedal, parénquima hepático aumentado de tamaño de aspecto granular fino. A nivel de mesenterio, marcado aumento de la ecogenicidad heterogénea, predominio hiperecoico, con áreas hipoecoicas o anecoicas. Vasos mesentéricos engrosados con paredes ecogénicas y luz visible. Presencia de múltiples adenopatías de aspecto inflamatorio y moderada cantidad de líquido libre en cavidad.
Es
ingresado en sala de gastroenterología, se le realizó tomografía
axial computarizada de abdomen con contraste oral y endovenoso: Aumento de la
densidad de la grasa mesentérica valores de -A0 UH. Área de tejido
de partes blandas que rodea los vasos mesentéricos superiores sin desplazarlos,
respeta la grasa adyacente, engrosamiento de la grasa perivascular, presencia
de adenopatías mesentéricas. Los hallazgos ecográficos
y tomográficos compatibles con paniculitis mesentérica. Se sugiere
la realización de biopsia de mesenterio.
Panendoscopia Oral que diagnosticó duodenitis eritematosa nodular.
Se interconsulta con cirugía para coordinar de biopsia de mesenterio, se realiza laparotomía exploratoria, en la inspección macroscópica se observó un engrosamiento difuso del mesenterio con pérdida de la consistencia de la grasa habitual con textura cérea, coloración anormal marrón-amarillento con áreas traslúcidas, engrosamiento y rigidez de los vasos mesentéricos, superficie de corte homogénea, sin quistes ni necrosis. Se realizó biopsia de mesenterio, que informó depósito de tejido rosado amorfo acelular, se realizó coloración de rojo congo donde se observó dicrolsmo positivo color verde manzana bajo luz polarizada y color rojo naranja intenso bajo de vasos sanguíneos (Fig. A, B y C).
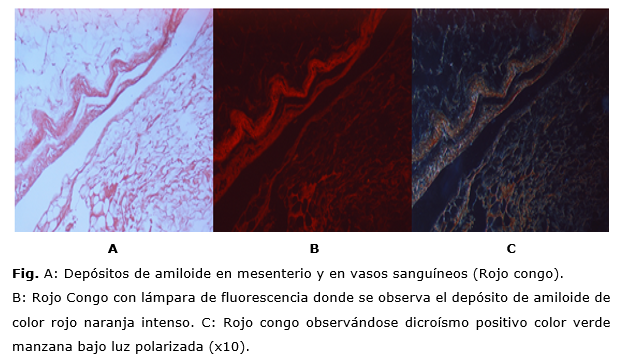
Se interconsulta con Hematología, Nefrología y Cardiología para la tipificación de la amiloidosis y se le brinda atención multidisciplinaria con instauración de tratamiento personalizado y evolución satisfactoria.
DISCUSIÓN
La amiloidosis incluye un grupo de trastornos dispares caracterizados por el depósito extracelular de fibrillas insolubles compuestas por proteínas agrupadas de forma irregular. Estas proteínas pueden acumularse en un área y provocar pocos síntomas o comprometer varios órganos y causar insuficiencias multiorgánica grave. La amiloidosis puede presentarse de novo o ser secundaria a varias infecciones, trastornos inflamatorios o enfermedades malignas.(6)
Las fibrillas de amiloide se componen de proteínas mal plegadas con solubilidad normal que se agregan en oligómeros y luego se convierten en fibrillas insolubres. Una serie de proteínas normales (de tipo salvaje) y mutantes son susceptibles de presentar ese plegamiento y agregación anormales (proteínas amiloidogénicas), lo que explica la gran variedad de causas y tipos de amiloidosis. Los depósitos de amiloide se tiñen de rosa con hematoxilina y eosina, se tiñen con ácido peryódico Shiff o con azul Alcián, pero lo característico es su birrefringencia verde manzana con microscopia de luz polarizada después de la tinción con rojo Congo.(6)
Para desarrollar amiloidosis, además de la producción de las proteínas amiloidogénicas, es probable un fracaso de los mecanismos de eliminación normales para este tipo de proteínas, se acumula y los depósitos de amiloide interfieren con la estructura y el funcionamiento de los órganos.(7)
Los principales tipos sistémicos de amiloidosis son:
En la amiloidosis primaria, el plegado anormal es el resultado de un evento proteolítico o una secuencia aminoacídica que da una cadena ligera inestable y lleva a su autoagregación. Los agregados forman protofilamentos que se asocian dentro de la proteína fibrilar.(8) En todos los tipos de amiloidosis los glucosaminoglicanos, mitades de los proteoglicanos y el amiloide P sérico (SAP)(9) interactúan con las fibrillas amiloides, promueven la formación de fibrillas y la estabilidad en los tejidos. La disfunción de un órgano resulta de una disrupción de la arquitectura del tejido por los depósitos de amiloide.(10)
En la amiloidosis primaria la proteína fibrilar está constituida por cadenas ligeras de inmunoglobulina (con mayor frecuencia lambda que kappa en una relación 2:1). Es el tipo de amiloidosis que se observa en las discrasias de células plasmáticas, mieloma múltiple y macroglobulinemia.(4)
La amiloidosis primaria es caracterizada por depósitos de fibrillas amiloides en el corazón, ocasiona falla cardíaca congestiva o arritmias; riñón, produce síndrome nefrótico o falla renal; el hígado, causa hepatomegalia y falla hepática colestásica progresiva; o el sistema neurológico, se manifiesta como neuropatía periférica sensoriomotora y disfunción autonómica.(7)
La hepatomegalia es común y puede ocurrir como resultado de congestión por falla cardíaca derecha o infiltración de amiloide en el hígado. La hepatomegalia de la infiltración amiloide puede ser masiva y al examen físico el hígado suele ser como una roca dura y poco suave. La profunda elevación de la fosfatasa alcalina con ligera elevación de transaminasas es característica de la amiloidosis hepática porque la infiltración ocurre en sinusoides. El compromiso del tracto gastrointestinal puede ser focal o difuso y los síntomas dependen del sitio y de la extensión. La macroglosia y la artritis temporo - mandibular producen xialorrea y dificultad en la masticación. La dismotilidad intestinal puede provocar diarrea, estreñimiento, megacolon, incontinencia fecal o prolapso rectal. Puede generar seudoobstrucción intestinal y obstrucción mecánica por infiltración amiloidótica del mesenterio.(11)
La malabsorción puede ser consecuencia del sobrecrecimiento bacteriano, o de la deposición amiloidea submucosa que genera una barrera física para la absorción. La obstrucción en la salida del estómago, funcional o mecánica, puede deberse a dismotilidad o presencia de un amiloidoma antral. Se puede observar hemorragia digestiva, infarto intestinal y enteropatía perdedora de proteína.(10)
Los hallazgos principales en las radiografías de intestino delgado son engrosamiento de las válvulas conniventes, dilatación del intestino por reemplazo de las capas muscular y presencia de numerosas lesiones nodulares. Los estudios por imágenes del colon pueden mostrar defectos de lleno, úlceras secundarias a la isquemia o estenosis y rigidez, es especial en el colon sigmoide y recto. La hemorragia es una importante manifestación y puede ser un serio problema. La manifestación común del sangrado es la púrpura debido a la fragilidad vascular y como resultado del depósito de amiloide en el endotelio y puede ser una amenaza para la vida posterior a una biopsia hepática o renal. La púrpura periorbitaria (ojos de mapache) es característica.(12)
La evaluación del grado de depósito amiloide es importante porque ayuda al pronóstico, plan terapéutico y determina la respuesta al tratamiento. El examen físico y la biopsia de los tejidos son los métodos para determinar el compromiso de un órgano.(12)
La
terapéutica actual para la amiloidosis sistémica está basada
en que la disfunción de un órgano mejora e incrementa la supervivencia
si la síntesis del precursor de la proteína amiloidogénica
es detenida. Por lo tanto, la principal terapia en la amiloidosis primaria es
reducir la fuente de cadenas ligeras monoclonales y suprimir la discrasia de
células plasmáticas subyacentes. La decisión sobre un régimen
de tratamiento específico para pacientes individuales debe ser tomada
en consideración en un equilibrio entre la eficacia del tratamiento anticipado
y la tolerabilidad.
La amiloidosis es un diagnóstico infrecuente, que no existe un protocolo de tratamiento de aceptación universal, requiere de una atención médica multidisciplinaria con un tratamiento específico y personalizado en dependencia de la extensión y estadío evolutivo de la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1.
Gertz MA. Diagnosing Primary Amyloidosis. Mayo Clinic Proceedings. [Internet]
2002;77(12):1278-1279. [acceso: 18/12/2025]. Disponible en: https://doi.org/10.4065/77.12.1278
2.
Lain Entralgo P. Historia de la medicina moderna y contemporánea. 2da
ed. Barcelona: Editorial Científico-Médica; 1963.
3.
Muchtar E, Grogan M, aus dem Siepen F, Waddington Cruz M, Misumi Y, Carroll
AS, et al. Supportive care for systemic amyloidosis: International Society of
Amyloidosis (ISA) expert panel guidelines. Amyloid. [Internet]. 2025;32(2):93-116.
[acceso: 18/12/2025]. Disponible en: https://doi.org/10.1080/13506129.2025.2463678
4.
Miller DV, Bhatt KN, Bocsi GT, Chang A, Gopal PP, Hernandez M, et al. Laboratory
Workup of Amyloidosis. Archives of Pathology & Laboratory Medicine. [Internet]
2026;150(3):179-193. [acceso: 18/12/2025]. Disponible en: https://doi.org/10.5858/arpa.2025-0275-CP
5.
Vishal Kukreti, Matthew D Seftel, Maria Adela Aguirre, Muayad Azzam, Deborah
Boedicker, Naresh Bumma, et al. American Society of Hematology (ASH) 2026 Guidelines
on Diagnosis of Light Chain Amyloidosis. Blood Advances. [Internet] 2026;017073.
[acceso: 18/12/2025]. Disponible en: https://doi.org/10.1182/bloodadvances.2025017073
6.
Tan M, Chen Y, Ooi M, de Mel S, Tan D, Soekojo C, et al. AL amyloidosis: Singapore
Myeloma Study Group consensus guidelines on diagnosis, treatment and management.
Annals of the Academy of Medicine, Singapore. [Internet]2023;52(11):601-624.
[acceso: 28/01/2026]. Disponible en: https://doi.org/10.47102/annals-acadmedsg.2023101
7.
Bucurica S, Nancoff AS, Moraru MV, Bucurica A, Socol C, Balaban DV, et al. Digestive
Amyloidosis Trends: Clinical, Pathological, and Imaging Characteristics. Biomedicines.
[Internet] 2024;12(11):2630. [acceso: 28/01/2026]. Disponible en: https://doi.org/10.3390/biomedicines12112630
8.
Rieger MJ, Flammer AJ, Gerull S, Pabst T, Auner HW, Samii K, et al. Updated
recommendations for the treatment of light-chain amyloidosis from the Swiss
Amyloidosis Network. Swiss Medical Weekly. [Internet] 2025;155:4219. [acceso:
28/01/2026]. Disponible en: https://doi.org/10.57187/s.4219
9.
Wu YW, Wei CH, Lin YH, Huang SY, Liu YW, Tan TD, et al. Joint position statement
of the diagnosis and management of light chain amyloidosis and light chain cardiac
amyloidosis by the Taiwan Society of Cardiology (TSOC) and the Hematology Society
of Taiwan (HST). Journal of the Formosan Medical Association.[Internet] 2025.
[acceso: 28/01/2026]. Disponible en: https://doi.org/10.1016/j.jfma.2025.05.006
10.
Malone MAV, Castillo DAA, Santos HT, Kaur A, Elrafei T, Steinberg L, et al.
A systematic review of the literature on localized gastrointestinal tract amyloidosis:
Presentation, management and outcomes. European Journal of Haematology. [Internet]2024;113(4):400-415.
[acceso:28/01/2026]. Disponible en: https://doi.org/10.1111/ejh.14269
11.
Rietz M, Weber T, Schaller T, Luitjens JH, Uhrmacher L, Messmann H, et al. Severe
gastroparesis complicated by gastric perforation caused by light-chain amyloidosis.
Zeitschriftfür Gastroenterologie. [Internet] 2025;63(4):387-392. [acceso:
28/01/2026]. Disponible en: https://www.thieme-connect.com/products/ejournals/abstract/10.1055/a-2442-7944
12.
Arslan DF, Inal E, Isik EG, Sanli Y. Extending Amyloidosis Diagnosis: A Rare
Case of Peritoneal AL Amyloidosis Revealed by 18F-FDG PET/CT and 99mTc-PYP SPECT/CT.
Clinical Nuclear Medicine.[Internet]2025;50(3):e192-e194. [acceso: 28/01/2026].
Disponible en: https://journals.lww.com/nuclearmed/abstract/2025/03000/extending_amyloidosis_diagnosis__a_rare_case_of.46.aspx
Conflicto de interés
Los autores declaran que no existen conflictos de interés.
Financiación
Los autores declaran que no hubo financiación involucrada en este trabajo.
Contribución de los autores
Conceptualización:
José Fernández Sotolongo, Yosvani Josué Ortiz Montero,
Licet González Fabián.
Curación
de datos: José Fernández Sotolongo.
Investigación:
José Fernández Sotolongo, Yosvani Josué Ortiz Montero,
Miguel Bernal Duran, Licet González Fabián.
Metodología:
Miguel Bernal Duran.
Administración
del proyecto: José Fernández Sotolongo.
Recursos:
Miguel Bernal Duran, Licet González Fabián.
Software:
Yosvani Josué Ortiz Montero.
Validación:
Yosvani Josué Ortiz Montero.
Redacción-
Elaboración del borrador original: José Fernández Sotolongo.
Redacción-
Revisión y edición: Yosvani Josué Ortiz Montero.